To barn døde etter å ha fått genterapi mot spinal muskelatrofi
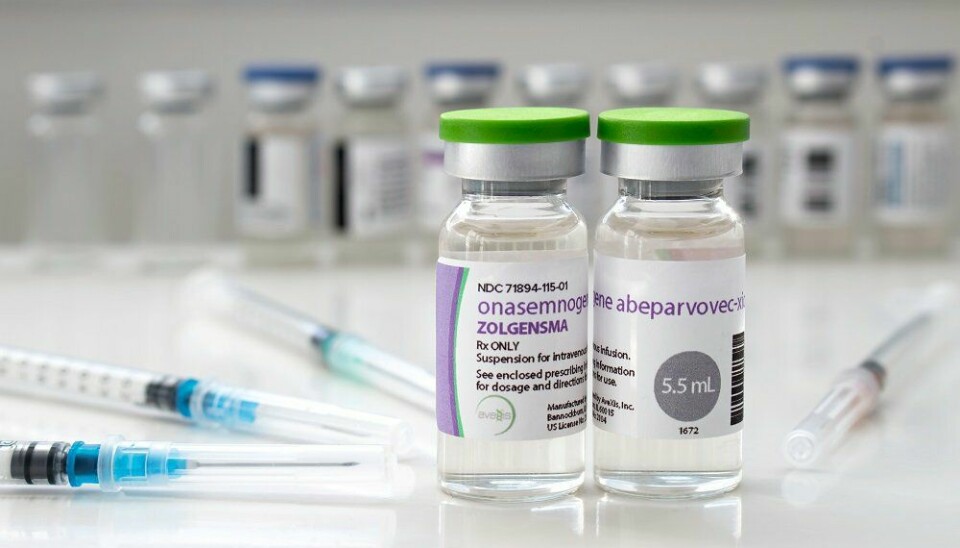
To barn som fikk genterapien Zolgensma mot spinal muskelatrofi (SMA), er døde av leversvikt, opplyser produsenten Novartis.
Barna døde fem til seks uker etter å ha fått Zolgensma – en form for genterapi, som er blitt omtalt som «verdens dyreste legemiddel» – opplyser det sveitsiske legemiddelfirmaet.
Annonse kun for helsepersonell
– Selv om akutt leversvikt er en kjent bivirkning, er dette de første tilfellene med dødelig utfall, opplyser Novartis. De sier de vil oppgradere pakningsvedlegget og spesifisere at det er meldt om dødsfall som følge av leversvikt.
De to barna døde i Russland og Kasakhstan. De to landenes helsemyndigheter er varslet. Det samme er andre land der Zolgensma brukes, skriver produsenten i en epost.
I bruk i Norge
Annonse kun for helsepersonell
I oktober i fjor bestemte Beslutningsforum at legemiddelet skulle tilbys i Norge. Behandlingen, som er designet for å ha livslang virkning, skal tas før symptomer oppstår, og det er derfor først og fremst nyfødte som vil få tilbudet.
Vedtaket er historisk, for dette var første gang Beslutningsforum sier ja til en avtale som innebærer at produsenten Novartis må tilbakebetale et betydelig beløp dersom barna som behandles med Zolgensma dør eller trenger permanent pustehjelp.
SMA er en alvorlig, sjelden sykdom som forårsaker muskelsvinn. Årsaken er en genfeil. I Norge antas det at cirka 50 personer under 18 år har SMA, mens tallet for personer over 18 år ikke er kjent, ifølge Helsenorge.no. I Europa og USA anslås SMA å opptre ved henholdsvis 1 av 4.000 og 1 av 10.000 fødsler, ifølge NORD, en amerikansk nasjonalforening for sjeldne sykdommer.
Zolgensma er en engangsbehandling som er designet for å ha livslang effekt. Om lag 7 pasienter diagnostiseres årlig med SMA i Norge og om lag fire til seks pasienter er aktuelle for behandling med Zolgensma.
Slik virker Zolgensma
Zolgensma er en genterapi til behandling av spinal muskelatrofi (SMA). Ved spinal muskelatrofi dør en type nerveceller i ryggraden gradvis. Disse cellene kalles motornevroner og har som oppgave å gi signaler fra hjernen til musklene. Når motornevronene gradvis dør, vil også musklene gradvis bli borte fordi nervesystemet ikke lenger har kontakt med dem. En feil i genet SMN1 gjør at det lages for få virksomme proteiner som kan passe på disse cellene. Proteinene heter på engelsk «Survival Motor Nevron», forkortet til «SMN-proteiner», og proteinene hjelper altså motornevronene med å overleve. Zolgensma er en erstatning for SMN1-genet og gis en gang som infusjon til en blodåre i armen. Tilførsel av genet kan stoppe ytterligere tap av motornevroner, men behandlingen kan ikke gjenopplive døde nevroner.
Den europeiske legemiddelmyndigheten (EMA) har kun godkjent at Zolgensma brukes til å behandle pasienter med to eller tre kopier av SMN2-genet og/eller pasienter med SMA type 1. Nytten av Zolgensma er undersøkt blant pasienter med symptomer på SMA type 1.
Pasientene hadde fått symptomer på sykdommen før de fylte seks måneder og hadde to kopigener av SMN2. De fleste pasientene som fikk Zolgensma i kliniske studier, var i live ved 18 måneders alder og etter 24 måneders oppfølging, uten at de trengte permanent støtte for å puste. Flere av barna som ble behandlet med Zolgensma, kunne dessuten sitte selvstendig, og noen klarte også etter hvert å stå eller gå med eller uten støtte.
Det er også vist at nytten av å starte behandling like etter fødsel, det vil si før barnet har utviklet symptomer på SMA, er stor. Nyfødtscreening av SMN1-genet kan avdekke SMA før symptomer oppstår, slik screening er fra høsten 2021 innført i Norge. For både Zolgensma og Spinraza gjelder at jo yngre pasienter med to eller tre kopigener er når de mottar behandlingen, desto større er effekten av behandlingen.
Kilde: Legemiddelverket