Legemidler og biotek
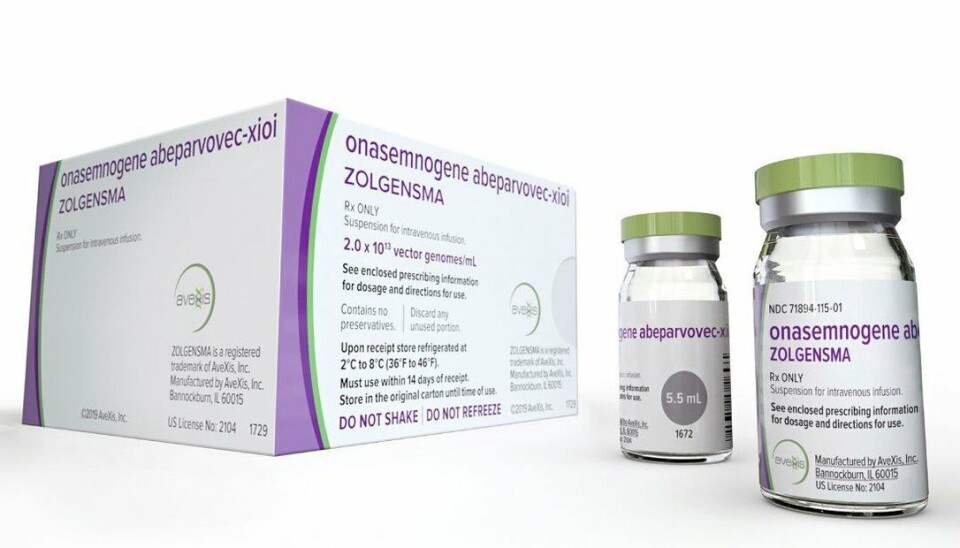
20 millioner kroner for en kur: Bestillerforum sier nei til hurtig metodevurdering av genterapien Zolgensma
Bestillerforum sier nei til å starte en hurtig metodevurdering av genterapien Zolgensma til behandling av spedbarn med spinal muskelatrofi (SMA). Begrunnelsen er at Novartis ikke har levert tilstrekkelig dokumentasjon til at Statens legemiddelverk kan utarbeide en kostnad-nytte-vurdering. Dette betyr at en innføring av genterapibehandlingen er utsatt på ubestemt tid. - Vi har sendt inn omfattende og relevant dokumentasjon, sier Sissel Rodahl i Novartis Gene Therapies
Spinal muskelatrofi (SMA) hører til en gruppe sykdommer som er kjennetegnet ved at de motoriske cellene i ryggmargen ødelegges. Dette er den vanligste dødelige og arvelige sykdommen hos barn i vår del av verden. Det har i gjennomsnitt vært diagnostisert syv nye SMA-pasienter per år de siste årene i Norge. Zolgensma (Onasemnogene abeparvovec) bruker et virus for å gi en normal kopi av SMN1-genet til babyer som er født med et defekt gen.
Setter en ny strandard
Zolgensma som er en engangsbehandling har en listepris i USA på 2,1 millioner dollar eller nesten 20 millioner kroner, og er med dette verdens dyreste legemiddel. Legemidlet er utviklet for å være en kur for SMA. Men den svært høye engangsprisen kombinert med kliniske data som gjør det utfordrende for Statens legemiddelverk å gjennomføre en kostnads-nytte analyse, gjør det krevende for Zolgensma å komme gjennom det norske beslutningssystemet for sykehuslegemidler.
Prinsipiell og konsekvensrik beslutning
Beslutningen er prinsipielt viktig, for nå kommer det snart legemidler basert på celle- og genterapi innen flere diagnoser - blant annet kreft og hemofili - som kan kurere alvorlige kroniske sykdommer med bare én behandling, og pasientene slipper livslang medisinering og legebesøk. Prislappen på engangsbehandlingene kan bli 20 millioner kroner per pasient. Oppfølgingen i studier av disse nye legemidlene er ofte korte, i blant bare et par år. Spørsmålet som legemiddelmyndighetene stiller seg er om de er sikre og helbredende, eller kommer sykdommen tilbake?
Prislappen er satt med en forutsetning at legemidlet er kurerende, men det er ingen som med sikkerhet kan si at det vil ha effekt livet ut. Det gjør at både industrien og myndighetene nå stiller seg spørsmålet om hvem som skal bære risikoen om legemidlene ikke holder det de lover - er det skattebetalerne eller selskapene?
Omgjorde vedtaket
Bestillerforum er en del av Nye metoder, og avgjør hvilke nye legemidler som skal vurderes nærmere for eventuell innføring som rutinemessig behandling på nasjonalt nivå i den norske spesialisthelsetjenesten og hvordan dette skje, samt hvilken type metodevurdering som skal utføres. Selve vurderingen av de nye metodene gjennomføres av Statens legemiddelverk eller Folkehelseinstituttet. Når de nye metodene er faglig vurdert, legges det fram for endelig beslutning i Beslutningsforum.
På sitt møte den 28. januar 2019, besluttet Bestillerforum at Legemiddelverket skulle igangsette en hurtig metodevurdering. Men på sitt møte 26. oktober i år snudde Bestillerforum og besluttet at legemiddelverket i stedet skal nøye seg med å lage en forenklet metodevurdering som bare oppsummerer dokumentasjonen som foreligger. I tillegg skal Sykehusinnkjøp HF utarbeide et prisnotat - og ikke starte prisforhandlinger med Novartis, som er den vanlige prosedyren.
Kan ikke utarbeide en troverdig kost-nytte-analyse
Enhetsleder Ellen Nilsen i sekretariatet for Nye metoder forteller til HealthTalk at årsaken til snuoperasjonen er at Novartis ikke har levert inn tilstrekkelig dokumentasjon:
- Bestillerforum ba i første omgang [28. januar 2019, red.anm] om en hurtig metodevurdering med en kostnad-nytte-vurdering. Det viser seg imidlertid at firmaet så langt ikke har levert dokumentasjon som er tilstrekkelig til at Legemiddelverket kan utføre en slik metodevurdering. Derfor har Bestillerforum bedt om at det gjøres en forenklet metodevurdering som oppsummerer dokumentasjonen som foreligger, sier Ellen Nilsen.
Bestillerforum mener at de kliniske studiene som foreligger ikke er tilstrekkelige til at det kan utarbeides en troverdig IKER (Inkrementell Kostnads-Effekt Rate) i en kost-nytte-analyse. Et hovedproblem sett fra Bestillerforums side er at det ikke foreligger direkte sammenlignende studier som kan kvantifisere relativ effekt i forhold til relevant komparator det vil si et annet relevant legemiddel.
For at et legemiddel skal innføres i spesialisthelsetjenesten må Beslutningsforum fatte et positivt vedtak. Det gjøres på grunnlag av en hurtig eller fullstendig metodevurdering. I tillegg kommer også vurderinger knyttet til pris etter notat fra Sykehusinnkjøp. Metodevurdering og prisnotat inngår som et grunnlag for en samlet vurdering, hvor også skjønnsmessige vurderinger skal inngå. Skjønnsmessige vurderinger er først og fremst knyttet til vurderinger av kvalitet og usikkerhet i dokumentasjonen, og til budsjettkonsekvenser.
Det er i prinsippet ikke noe som hindrer Beslutningsforum å innføre et legemiddel basert på en forenklet metodevurdering, men det regnes som usannsynlig at en forenklet metodevurdering er tilstrekkelig i dette tilfellet. Det skyldes blant annet legemidlets høye pris og utfordringen med å utarbeide en troverdig IKER.
På spørsmål om en forenklet metodevurdering er et tilstrekkelig for at Beslutningsforum kan innføre Zolgensma svarer Ellen Nilsen. - Beslutningsforum for nye metoder fatter sine beslutninger basert på det beste tilgjengelig dokumentasjonsgrunnlaget på beslutningstidspunktet, og vurderer selv om beslutningsgrunnlaget er tilstrekkelig, forteller hun.
Novartis: - Vi har sendt inn omfattende dokumentasjon
Sissel Rodahl er daglig leder i Novartis Gene Therapies for området European Mid-Sized Countries. Hun forteller til HealthTalk at de har sendt inn omfattende dokumentasjon til Statens legemiddelverk og at de inviterer til dialog og nytenkning med Legemiddelverket og Sykehusinnkjøp om hvordan man skal dokumentere effekt og varighet av behandlingen og hvordan man skal bli enige om en bærekraftige pris- og betalingsmodeller:
- Vi har sendt inn omfattende og relevant dokumentasjon i henhold til den første forespørselen fra Statens Legemiddelverk, sier hun.
- Det vil alltid være områder med usikkerhet, spesielt for medisiner som Zolgensma, hvor én enkelt dose kan gi livslang effekt for sjeldne og alvorlige tilstander. Det er svært få pasienter i hver pasientgruppe, noe som medfører at de tradisjonelle metodene for å vurdere helsegevinsten ikke blir så lett å bruke. Dette krever at vi sammen med myndighetene tenker nytt, både om hvordan effekt og varighet av behandling skal dokumenteres, samt innføring av bærekraftige pris- og betalingsmodeller, sier Rodahl.
Har god dialog
- Vi har god dialog med både Statens legemiddelverk og Sykehusinnkjøp om dette. Vårt mål er at vi sammen med myndighetene skal finne løsninger som sikrer denne lille og sårbare pasientgruppen raskest mulig tilgang til behandling, forteller Sissel Rodahl.
SMA-pasientene får i dag Spinraza
I februar 2018 innførte Beslutningsforum Spinraza (nusinersen) ved SMA på gitte vilkår som i praksis er tilfredsstilt hos de fleste barn med nydiagnostisert SMA. Det er legemiddelselskapet Biogen som har utviklet Spinraza
Hver dose med Spinraza koster i USA om lag én million norske kroner. Barn med SMA trenger syv sprøyter det første året, og deretter tre sprøyter med medisinen resten av livet.
Prisene som staten betaler for sykehuslegemidler er konfidensielle. Hovedargumentet for hemmelighold av priser er at det gir legemiddelselskapene mulighet til å gi høye rabatter til land som Norge uten at andre land vet hva vi betaler.
Etter prisforhandlinger mellom Biogen og Sykehusinnkjøp er den norske prisen på Spinraza etter det HealthTalk erfarer i størrelsesorden 500 000 kroner per dose.
I USA er som nevnt prisen på Zolgensma i underkant av 20 millioner kroner. Novartis begrunner den høye prisen med at den har bedre effekt og at den samlet sett er billigere enn Spinraza som altså er en livslang behandling.
Det sentrale spørsmålet er om denne prisen blir akseptert i Norge, eller om Novartis må redusere prisen på Zolgensma betydelig i forhandlingene med Sykehusinnkjøp for at Beslutningsforum skal velge å innføre genterapien. Men for at slike forhandlinger skal starte må Legemiddelverket gjennomføre en hurtig metodevurdering, og om eller når det vil skje er det ingen i dag som vet.
SMA-konkurranse
De amerikanske legemiddelmyndighetene (FDA) godkjente Zolgensma i mai 2019. Grunnlaget for godkjenningen var den innledende studien publisert i The New England Journal of Medicine som viste svært lovende effekter. Resultatene opprettholdes i oppfølgingsstudier.
Det europeiske legemiddelverket (EMA) sitt ekspertpanel for godkjenning av legemidler (CHMP) ga i mai i år en betinget godkjenning av Zolgensma for visse pasienter: De med type 1 SMA, den alvorligste formen for sykdommen (hvor den høyeste motoriske funksjon er liggende) og for SMA-pasienter med opptil tre kopier av SMN2-genet, en indikator på sykdommens alvorlighetsgrad.
EU-Kommisjonen har det endelige ordet når det gjelder godkjenning av nye legemidler, men Kommisjonen følger normalt EMAs anbefaling. Normalt kommer en slik godkjenning et par måneder etter CHMP har avgitt sin innstilling, og forsinkelsen betyr trolig at EU-kommisjonen trenger å gjøre ytterligere vurderinger før de konkluderer.
Zolgensma er altså den andre behandlingen for SMA som ligger an til å få en EU-godkjenning etter Biogens Spinraza.
EU holder i disse dager på å vurdere et tredje SMA-legemiddel. Det er risdiplam fra Roche. Medisinsk direktør Karsten Bruins Slot i Roche Norge sier at de forventer at risdiplam blir godkjent i EU tidlig neste år. Det vil i så fall bety at risdiplam kan komme opp til vedtak i Beslutningsforum før sommeren 2021 .
Risdiplam er studert i et bredt klinisk utviklingsprogram i SMA, med pasienter fra fødsel til 60-års alder, og inkluderer også pasienter tidligere behandlet med andre SMA-behandlinger. Studiepopulasjonen representerer et bredt spekter av mennesker som lever med sykdommen. Risdiplam studeres i totalt fire studier: FIREFISH (SMA type 1) og SUNFISH (SMA type 2 og 3) er de to studiene som danner grunnlaget for søknaden om markedsføringstillatelse. I tillegg studeres risdiplam i studiene JEWELFISH (pasienter som tidligere har mottatt annen behandling) og RAINBOWFISH (nyfødte barn med genetisk påvist risiko for SMA, uten symptomer).
Risdiplam administreres oralt (gjennom munnen eller i ernæringssonde) som en flytende væske. Risdiplam er designet for å gi en vedvarende økning samt å bevare SMN-proteinnivået i sentralnervesystemet og i perifere vev. Risdiplam har en virkningsmekanisme som gjør at SMN2-genet hjelpes til å produsere mer funksjonelt SMN-protein i hele kroppen.